
Take away computation from the world and everything is wrapped in blind ignorance. — Isidore of Seville (550-636)
Our work focuses on the theory of materials, especially disordered materials and the development of new methods to better understand and predict the outcomes of experiment. Scholargps summary here.





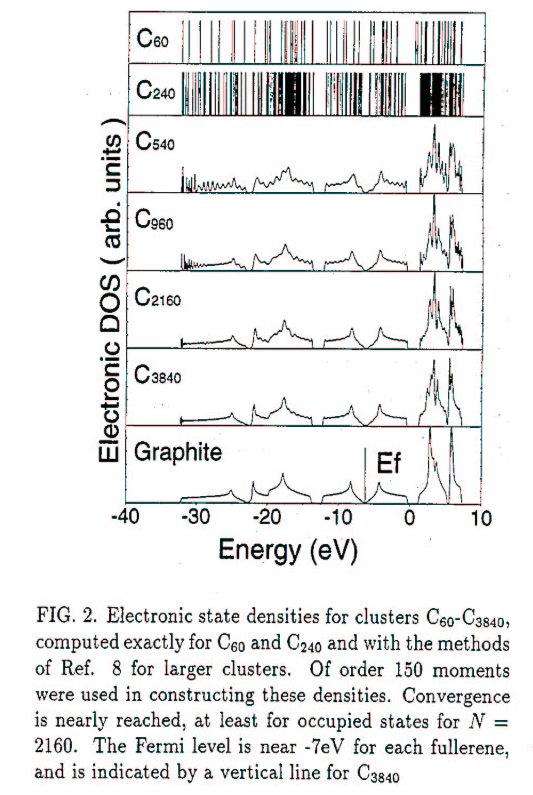


Our research was sustained by the National Science Foundation and the Army Research Office for many years, and currently by the generous support of the Department of Energy, Pacific Northwest National Labs, and the Office of Naval Research.
